Системная средиземноморская лихорадка
Системная средиземноморская лихорадка
Общая информация

Системная средиземноморская лихорадка (ССЛ) (Familial Mediterranean Fever, FMF) — аутовоспалительное заболевание, характеризующееся периодически повторяющимися приступами лихорадки в сочетании с асептическим перитонитом, иногда плевритом, поражением кожи, артритом и редко перикардитом1.
Эпидемиология

Среди всех аутовоспалительных заболеваний для ССЛ в наибольшей степени характерна этническая предрасположенность. Наиболее подвержены заболеванию представители четырех этносов: евреи-сефарды (североафриканские, неашкеназские евреи), турки, арабы и армяне1. Знание этнической принадлежности пациента оказывает существенную помощь при установлении диагноза.
Но по мере изучения ССЛ стало понятно, что распространенность ССЛ не ограничивается указанными выше популяциями. Благодаря миграции населения в ХХ веке, заболевание распространилось по всему миру: США, Европа, Россия, Япония, Южная Америка и др2. Таким образом, заболевание было выявлено у представителей достаточно отдаленных этнических групп, что предостерегает от постановки диагноза исключительно на основе родословной.
Этиология и патогенез
Этиологическим фактором ССЛ выступает мутация гена MEFV, локализованного на коротком плече 16-й пары хромосом. Тип наследования — аутосомно-рецессивный, соответственно, если оба родителя являются здоровыми гетерозиготами, вероятность развития заболевания у их потомства составляет 25% (рис. 1). Вместе с тем имеются данные о возможности аутосомно-доминантного типа наследования при наличии мутации с высокой пенетрантностью3.
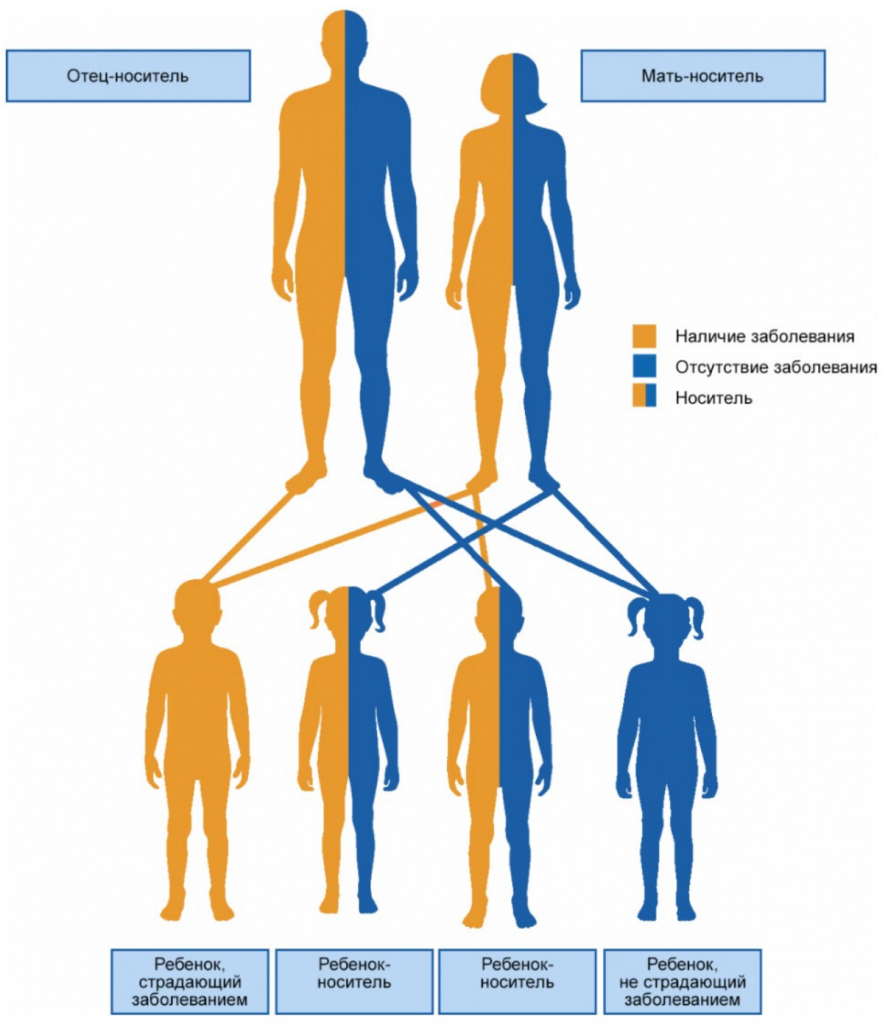
Рисунок 1. Аутосомно-рецессивный тип наследования
Мутации, ассоциированные с ССЛ, являются мутациями усиления функции, то есть они придают белку новую или повышенную активность с эффектом дозы аномального гена (т. е. увеличение количества копий аномального гена дает больший эффект). Ген MEFV в норме кодирует белок (так называемый пирин), который экспрессируется в циркулирующих нейтрофилах4.
Пирин представляет собой неотъемлемый структурный компонент врожденной иммунной системы и участвует в защитных реакциях от бактериальных токсинов, которые деполимеризуют актин и активируют инфламмасомы. Бактерии, которые производят подобные токсины, включают Clostridium difficile, Burkholderia cenocepacia, а также Vibrio cholerae4. Предполагается, что пирин притупляет воспалительный ответ, возможно, путем ингибирования хемотаксиса и активации нейтрофилов. Генные мутации приводят к появлению измененных молекул пирина, которые не ингибируют активацию инфламмасом.
Нарушение функции инфламмасом ведет к спонтанному запуску воспалительного каскада и гиперактивации системы врожденного иммунитета без видимой причины (в отсутствие инфекции или аутоиммунной реакции), основная патогенетическая роль в этом процессе принадлежит провоспалительному цитокину ИЛ-1β4,5.

Интерлейкин-1β активирует клетки врожденного иммунитета (нейтрофилы, моноциты; Т-лимфоциты; остеокласты), индуцирует экспрессию коллагеназ, разрушающих суставной хрящ, стимулирует выработку простагландина E2 в гипоталамусе, что приводит к развитию гипертермии центрального генеза. Воздействуя на эндотелий сосудов, ИЛ-1β способствует развитию сыпи и стимулирует атерогенез в кровеносных сосудах, а также активирует синтез острофазовых белков в печени5.
Клинические последствия гиперпродукции ИЛ-1β — спонтанные приступы системного воспаления с увеличением количества лейкоцитов за счет нейтрофилов и острофазовых показателей крови.
ССЛ дебютирует, как правило, до 20 лет. К основным типичным клиническим признакам заболевания относятся: лихорадка, абдоминальные проявления в виде острой боли в животе, артрит, плеврит, перикардит, сыпь на голенях и стопах, мышечные проявления и поражение мочеполовой системы3,6.
Клиническая картина
ССЛ характеризуется периодическими повторяющимися атаками системного воспаления продолжительностью 6–72 ч, проявляющегося фебрильной лихорадкой 38–40,5 °С и симптомами воспаления органов и систем6,7. Основные проявления заболевания представлены на рис. 2.
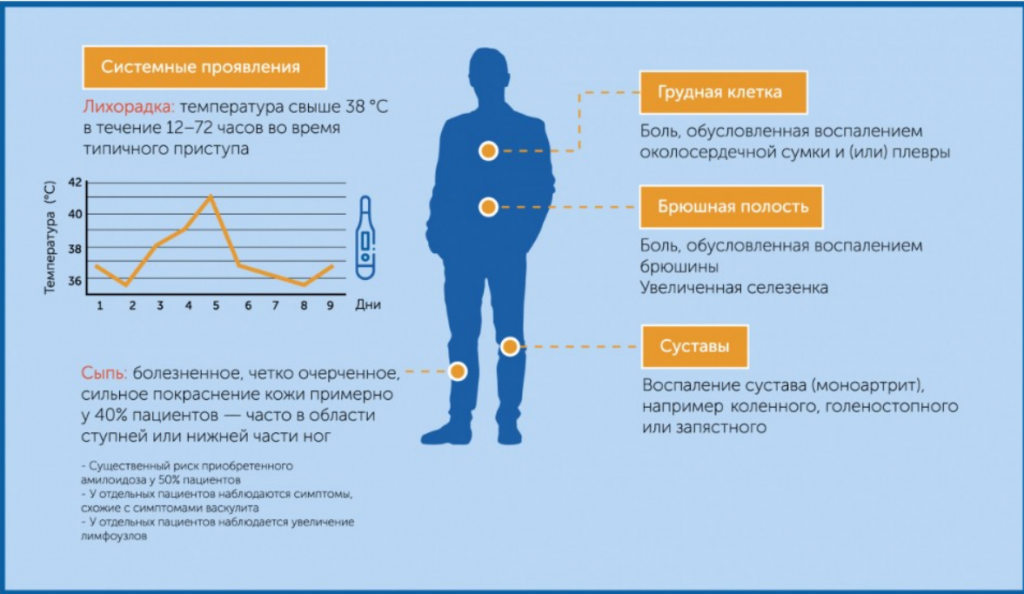
Рисунок 2. Клинические проявления ССЛ
Локализация процесса происходит преимущественно в четырех основных зонах3:
- Абдоминальная: с развитием асептического перитонита, проявляющегося выраженным болевым синдромом с картиной «острого живота».
- Торакальная: с развитием асептического плеврита, в большинстве случаев одностороннего.
- Суставная: с развитием артритов, главным образом суставов нижних конечностей.
- Кожная: основное проявление — рожеподобная эритема, как правило на голенях, над голеностопным суставом,
в котором имеются признаки артрита, и на тыльной поверхности стоп.
Частота атак ССЛ различна, что отражает ее тяжесть: от 1–2 раз в неделю до нескольких в год. Во время атаки возможны любые сочетания фебрильной лихорадки с проявлениями воспаления со стороны указанных систем. Редко атаки могут протекать в виде изолированной лихорадки. Практически всегда фебрильные атаки сопровождаются значительным повышением уровня острофазовых маркеров: СОЭ, СРБ, сывороточного амилоида А (SAA)8,9.
Значения острофазовых маркеров существенно снижаются вне атаки, но примерно у 63% пациентов сохраняется повышенный уровень того или иного острофазового показателя в промежутке между атаками. Ключевым и наиболее чувствительным маркером воспаления, в качестве оценки активности заболевания, эффективности лечения и риска развития амилоидоза, в настоящее время служит сывороточный амилоид А (SAA)3,10.
Существует три11 варианта течения (фенотипа) ССЛ12,13:
- ССЛ I типа: протекает в виде типичных атак.
- ССЛ II типа: первым клиническим проявлением данного фенотипа является амилоидоз почек. До развития амилоидоза
у таких пациентов можно обнаружить только повышение уровня острофазовых маркеров. - ССЛ II типа: первым клиническим проявлением данного фенотипа является амилоидоз почек. До развития амилоидоза у таких пациентов можно обнаружить только повышение уровня острофазовых маркеров.
Осложнения
Амилоидоз почек — основное осложнение, которое определяет прогноз заболевания и приводит к развитию хронической почечной недостаточности. Хроническая почечная недостаточность вследствие амилоидоза – причина смерти 90% больных 14 с FMF14. Частота амилоидоза составляет 25–30%1 и может зависеть от этнического происхождения пациента15. Так, у нелеченых пациентов турецкого происхождения частота амилоидоза достигает 60%, у евреев-сефардов — 75%15.
Факторы риска развития амилоидоза при ССЛ: мужской пол, наличие артрита, задержка диагноза, неадекватное лечение, а также семейный анамнез амилоидоза14.
Другим осложнением, имеющим существенно меньшее значение для прогноза заболевания, является спаечная болезнь.
Клиническими последствиями хронического воспаления при ССЛ могут быть нормоцитарно-нормохромная анемия, спленомегалия, задержка роста у детей, снижение плотности костей, мужское и женское бесплодие, преждевременные роды, повышенный риск сердечно-сосудистых заболеваний11.
Диагностика
Постановка диагноза ССЛ возможна на основании клинических данных, но для подтверждения диагноза важно провести молекулярно-генетическое исследование, которое особенно информативно при оценке атипичных случаев.
Подробнее о программе генетической диагностики.
Генетическое тестирование используется для подтверждения, но не для исключения диагноза периодической болезни. Встречаются 5 основных патогенных мутаций гена MEFV: M694V, M694I, M680I, V726A, E148Q. Наибольшее число мутаций (до 38%) приходится на 10-й экзон. Мутации M694V приводят к более тяжелому течению заболевания с ранним началом, частому поражению суставов, высокому уровню амилоидоза при отсутствии лечения. Пациенты, гомозиготные по мутации M694V, в 6 раз чаще страдают амилоидозом по сравнению с другими генотипами. Мутации обнаруживаются также в экзонах 2, 3, 516,17,18.
Следует иметь в виду, что у некоторых пациентов с очевидными фенотипическими признаками ССЛ выявляется только один мутантный ген, а иногда отсутствуют явные мутации в гене MEFV. Приблизительно у 10–20% пациентов, которые отвечают диагностическим критериям ССЛ, мутации в гене MEFV не выявляются, что предполагает наличие эпигенетических факторов и факторов внешней среды, способствующих патогенезу заболевания19. Неспецифические признаки включают: повышение уровня лейкоцитов с преобладанием нейтрофилов, СОЭ, С-реактивного белка и фибриногена. Экскреция с мочой > 0,5 г белка за сутки предполагает формирование амилоидоза почек19.
В диагностике ССЛ используют клинические диагностические критерии клиники Тель-Хашомер — это «золотой стандарт» (табл. 1)20.
Таблица 1. Диагностические критерии ССЛ клиники Тель-Хашомер20
Примечание.
Для постановки диагноза ССЛ необходимо соответствие ≥ 1 большим критериям, или ≥ 2 малым критериям, или 1 большому + ≥ 5 поддерживающим критериям, или 1 малому + ≥ 4 поддерживающим критериям из числа первых 5.
Лечение
Цель лечения ССЛ — достижение полного контроля над неспровоцированными приступами и минимизация субклинического воспаления в межприступный период21.
В настоящее время основное лечение ССЛ заключается в применении колхицина. Колхицин предотвращает атаки заболевания, снижает их частоту и тяжесть. Также важным фактором служит значительное уменьшение риска развития амилоидоза при применении адекватных доз препарата22.
Сегодня колхицин — единственный препарат, эффективность которого в отношении предотвращения амилоидоза доказана.
В соответствии с рекомендациями EULAR (2016) колхицин должен быть назначен сразу же после установления диагноза ССЛ (уровень доказательности А)22.
Колхицин эффективен у большинства, но далеко не у всех пациентов с ССЛ. У 20% пациентов с ССЛ на терапии колхицином
сохраняются воспалительные атаки и скрытое воспаление между приступами. Почему это происходит? Более 40% пациентов
не соблюдают рекомендации по лечению, 2–5% отмечают непереносимость и серьезные побочные эффекты терапии колхицином (тяжелая диарея, невропатия, рабдомиолиз и подавление активности костного мозга)23. Кроме того, существует понятие «колхицинрезистентности», когда пациент с ССЛ недостаточно отвечает на терапию максимально допустимой дозой колхицина. Колхицинрезистентность диагностируется при сохранении ≥ 1 атаки в месяц у комплаентных пациентов (получающих максимальную дозу колхицина ≥ 6 мес.) или при стойком повышении уровня СРБ/SАА в период между приступами21,24,25.
Доля пациентов с истинной колхицинрезистентностью достигает 15%. Таким пациентам показано назначение генно-инженерных биологических препаратов (ГИБП). Терапия ГИБП, блокирующими интерлейкин-1, — наиболее эффективный вариант патогенетического лечения пациентов с колхицинрезистентной ССЛ21. Выявление АА-амилоидоза требует интенсификации лечения с рассмотрением возможности биологической терапии вне зависимости от наличия
колхицинрезистентности у пациентов с ССЛ.
Одним из ингибиторов ИЛ-1 является канакинумаб — моноклональное антитело к ИЛ-1β. Канакинумаб селективно связывается с ИЛ-1β и полностью блокирует его действие. Препарат имеет длительный период полувыведения (26 дней), что делает возможным применять его при колхицинрезистентной ССЛ 1 раз в 4 недели в виде подкожных инъекций2
Эффективность и безопасность канакинумаба у пациентов с колхицинрезистентной ССЛ убедительно продемонстрированы в крупном рандомизированном двойном слепом плацебо-контролируемом исследовании CLUSTER (Canakinumab Pivotal Umbrella Study in Three Heredity Periodic Fevers)22,26. Канакинумаб — это единственный ГИБП, который зарегистрирован в Российской Федерации по показанию ССЛ (на 2021 год).
Заключение
ССЛ является самым распространенным моногенным аутовоспалительным заболеванием и имеет отчетливую этническую предрасположенность. В России проживает немало представителей этнических групп (армяне, азербайджанцы, народы Северного Кавказа), в которых отмечается высокая распространенность мутации гена MEFV и, соответственно, ССЛ. Благодаря внедрению в клиническую практику молекулярной генетической диагностики выявляемость ССЛ увеличивается.
Тяжесть и прогноз заболевания определяются не только атаками системного воспаления, но и высоким риском развития вторичного АА-амилоидоза, который может привести к развитию хронической почечной недостаточности и гибели пациента. Патогенетическая лекарственная терапия ГИБП позволяет достичь значительного прогресса в ведении таких пациентов с улучшением прогноза, предотвращением развития жизнеугрожающих осложнений и улучшением качества жизни.
Список литературы:
1. Салугина С.О., Федоров У.С., Агафонова Е.С. Моногенные аутовоспалительные заболевания детей и взрослых: что необходимо знать ревматологу. Научно-практическая ревматология. 2019;57(2):125-132. doi: 10.14412/1995-4484-2019-125-132
2. Ozen S., Frenkel J., Ruperto N. et al. The Eurofever project: towards better care for autoinflammatory disease. Eur J Pediatr. 2011;170(4):445-52.
3. Федоров Е.С., Салугина С.О., Кузьмина Е.Е. Семейная средиземноморская лихорадка (периодическая болезнь): современный взгляд на проблему. Современная ревматология. 2013; 1: 24-29.
4. Park Y.H., Wood G., Kastner D.L., Chae J.J.: Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17(8):914–921, 2016. doi: 10.1038/ni.3457
5. Lachmann HJ, et al. Arthritis Rheum. 2011;63:314-24.
6. Кузьмина Н.Н., Салугина С.О., Федоров Е.С. Аутовоспалительные заболевания и синдромы у детей: Учеб.-метод. Пособие. – М: ИМА-ПРЕСС, 2012. – 104 с.
7. Амарян Г.Г., Саркисян Т.Ф., Айрапетян А.С. Семейная средиземноморская лихорадка у детей (периодическая болезнь): клинические и генетические аспекты. Методическое пособие. Ереван; 2012.
8. Nir-Paz R., Ben-Chetrit E., Pikarsky E. et al. Unusual presentation of familial Mediterraneanfever: role of genetic diagnosis. Ann Rheum Dis. 2000.
9. Korkmaz C., Zdogan H., Kasapcopur О., Yazici H. Acute phase response in familial Mediterranean fever. Ann Rheum Dis. 2002;61(1):79-81.
10. Berkun Y., Padeh S., Reichman B. et al. A single testing of serum amyloid a levels as a tool for diagnosis and treatment dilemmas in familial Mediterranean fever. Semin Arthritis Rheum. 2007;37(3):182-8.
11. Ben-Zvi I, Livneh A. Nat Rev Rheumatol. 2011;7:105-12.
12. Hurwich B.J., Schwartz J., Goldfarb S. Record survival of siblingswith familial Mediterranean fever, phenotype 1 and 2. Arch Intern Med. 1970;125(2):308-11.
13.Kocak H., Besbas N., Saatci U., Bakkaloglu A. Amyloidosis in children with familial Mediterranean fever. Turk J Pediatr. 1989;31(4):281-7.
14. Kasifoglu T, Bilge SY, Sari I et al Amyloidosis and its related factors in Turkish patients with familial Mediterranean fever: a multicentre study. Rheumatology (Oxford) 2014; 53(4):741–745.
15. Shohat M., Magal N., Shohat T. et al. Phenotype-genotype correlation in familial Mediterranean fever: evidence for an association between Met694Val and amyloidosis. Eur J Hum Genet. 1999 Apr;7(3):287-92.
16. Yackov Berkun MD and Eli M. Eisenstein Update on Auto-Inflammatory Diseases and Familial Mediterranean Fever Isr Med Assoc J 2016;18(3-4):221-4.
17. The of hereditary auto-inflammatory disorders mutations. Infevers. https://infevers.umai-montpellier.fr/web/ доступно на 27.10.2020.
18. Grandmange S, et al. Genes Immun. 2011;12:497-503 5. Genes: MEFV. Genetics Home Reference. http://ghr.nlm.nih.gov/gene/MEFV доступно на 27.07.2021.
19. Booty M.G., Chae J.J., Masters S.L. et al: Familial Mediterranean fever with a single MEFV mutation: Where is the second hit? Arthritis Rheum 60(6):1851–1861, 2009. doi: 10.1002/art.24569
20. Sohar E., Gafni J. Tel Hashomer criteria for diagnosis of FMF. First International Conferens on FMF. London and Tel Aviv Freund Publishing Hause; 1997. 207 p.
21. S. Ozen et al. Ann Rheum Dis 2016; 75:644-651.
22. Demirkaya E., Erer B., Ozen S., BenChetrit E. Efficacy and safety of treatments in Familial Mediterranean Fever. Rheumatol Int. 2016;36(3):325-31.
doi: 10.1007/s00296-015-3408-9
23. Kacar M et al. J Inflamm Res 2020;13:141–149.
24. Рамеев В.В., Симонян А.Х., Богданова М.В. и др. Периодическая болезнь: эволюция представлений о заболевании и подходы к диагностике и лечению. Клин фармакол тер 2020;29(2):56-68.
25. Ben-Chetrit E., Amar S Clin Exp Rheumatol. 2008;27(2 Suppl. 53): S1-3.
26. Ozturk M.A., Kanbay M., Kasapoglu B. et al. Therapeutic approach to familial Mediterranean fever: a review update. Clin Exp Rheumatol. 2011;29(4 Suppl 67): S77-86.